Continuando con las técnicas de análisis de los productos lácteos, se expone seguidamente la metodología analítica para la determinación del índice de ácidos grasos volátiles solubles e insolubles en la mantequilla.
Principios y fundamentos metodológicos.
El índice de ácidos grasos volátiles solubles o índice de Reichert o Reichert-Meissl-Mellny, es el número de mililitros de una solución acuosa de álcali 0,1 N, requeridos para neutralizar los ácidos grasos volátiles solubles en agua de una cantidad de 5 gramos de grasa en las condiciones que se especifica en esta metodología.
El índice de ácidos grasos volátiles insolubles o índice de Polenske, es el número de mililitros de solución acuosa de álcali 0,1 N, requeridos para neutralizar los ácidos grasos volátiles insolubles en agua, obtenidos en una muestra de 5 gramos de grasa, en las condiciones del método.
Después de saponificar la grasa con una solución de sodio hidróxido en glicerina, la solución jabonosa se diluye con agua y se acidifica con ácido sulfúrico. Los ácidos grasos volátiles se destilan y los ácidos grasos insolubles se separan de los solubles por filtración. La solución acuosa de ácidos solubles y la solución etanólica de ácidos insolubles se valoran separadamente con una solución de álcali normalizada. El método es empírico porque sólo determina una parte de estos ácidos; por tanto, los aparatos utilizados y las especificaciones referentes al procedimiento se deben seguir rigurosamente para obtener resultados exactos y reproducibles.
Material y aparatos utilizados.
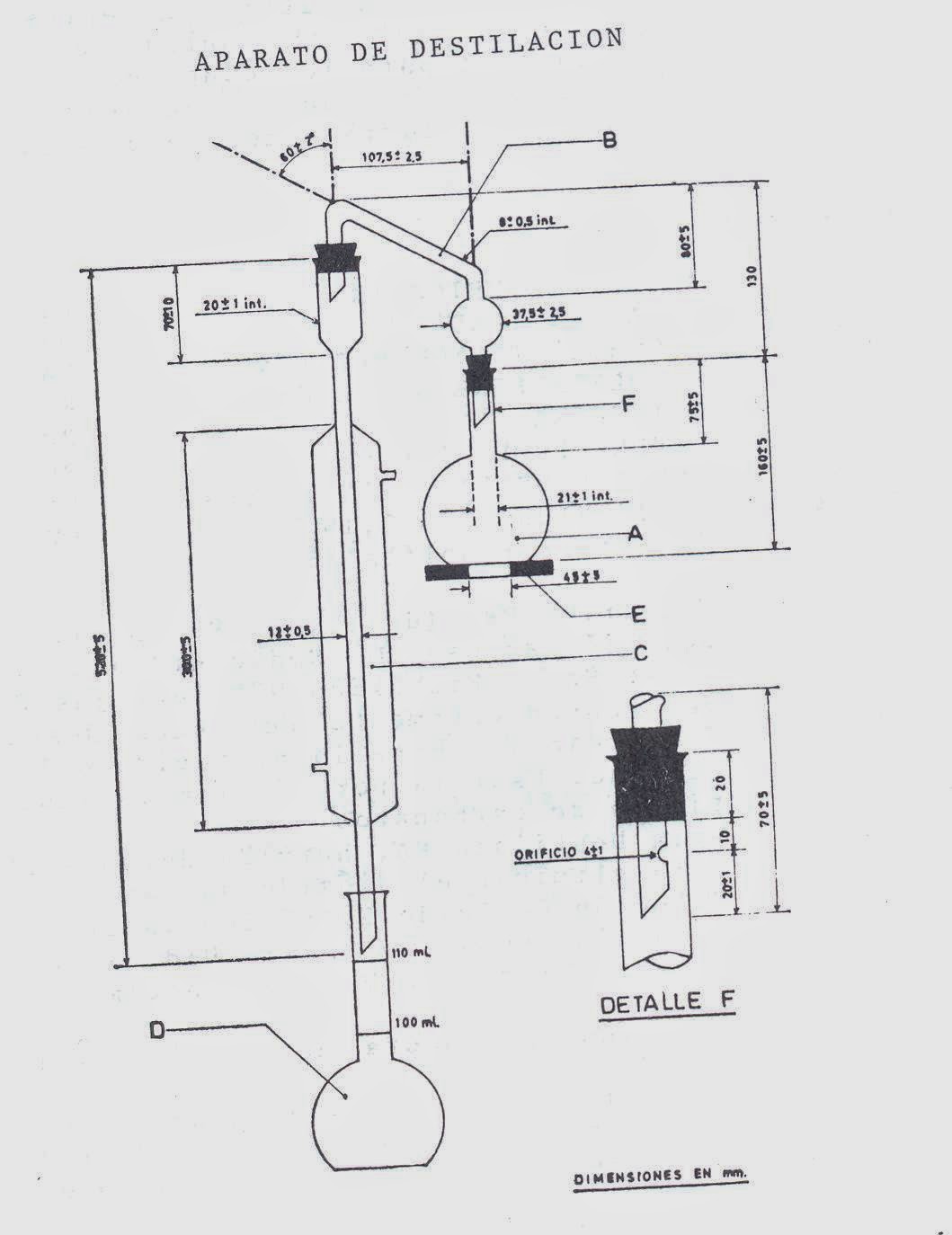
-Aparato de destilación (en esquema adjunto): Está integrado por los siguientes elementos:
1.Matraz de fondo plano de vidrio al borosilicato de 300 ml de capacidad (A).
2.Cabeza de destilación (E).
3.Refrigerante ( C).
4.Receptor, es un matraz aforado con las rayas circulares de aforo a 100 y 110 ml (D).
5.Lámina de amianto o asbesto de 120 mm de diámetro, 6 mm de espesor con una abertura central circular de 40 a 50 mm de diámetro, para sostener el matraz durante el calentamiento (E).
6.Piedra pómez triturada que pasa a través de un tamiz de malla circular de 1,44 mm.
En la figura se representan las dimensiones en mm y el montaje del aparato de destilación; para las conexiones se puede utilizar tapones de caucho, neopreno o silicona, o juntas de vidrio esmerilado “estandar” 24/40.
Principios y fundamentos metodológicos.
El índice de ácidos grasos volátiles solubles o índice de Reichert o Reichert-Meissl-Mellny, es el número de mililitros de una solución acuosa de álcali 0,1 N, requeridos para neutralizar los ácidos grasos volátiles solubles en agua de una cantidad de 5 gramos de grasa en las condiciones que se especifica en esta metodología.
El índice de ácidos grasos volátiles insolubles o índice de Polenske, es el número de mililitros de solución acuosa de álcali 0,1 N, requeridos para neutralizar los ácidos grasos volátiles insolubles en agua, obtenidos en una muestra de 5 gramos de grasa, en las condiciones del método.
Después de saponificar la grasa con una solución de sodio hidróxido en glicerina, la solución jabonosa se diluye con agua y se acidifica con ácido sulfúrico. Los ácidos grasos volátiles se destilan y los ácidos grasos insolubles se separan de los solubles por filtración. La solución acuosa de ácidos solubles y la solución etanólica de ácidos insolubles se valoran separadamente con una solución de álcali normalizada. El método es empírico porque sólo determina una parte de estos ácidos; por tanto, los aparatos utilizados y las especificaciones referentes al procedimiento se deben seguir rigurosamente para obtener resultados exactos y reproducibles.
Material y aparatos utilizados.
-Aparato de destilación (en esquema adjunto): Está integrado por los siguientes elementos:
1.Matraz de fondo plano de vidrio al borosilicato de 300 ml de capacidad (A).
2.Cabeza de destilación (E).
3.Refrigerante ( C).
4.Receptor, es un matraz aforado con las rayas circulares de aforo a 100 y 110 ml (D).
5.Lámina de amianto o asbesto de 120 mm de diámetro, 6 mm de espesor con una abertura central circular de 40 a 50 mm de diámetro, para sostener el matraz durante el calentamiento (E).
6.Piedra pómez triturada que pasa a través de un tamiz de malla circular de 1,44 mm.
En la figura se representan las dimensiones en mm y el montaje del aparato de destilación; para las conexiones se puede utilizar tapones de caucho, neopreno o silicona, o juntas de vidrio esmerilado “estandar” 24/40.
Reactivos necesarios.
-Ácido sulfúrico 1 N (SV).
-Agua destilada (PA).
-Alcohol etílico al 96% (v/v, PA).
-Fenolftaleína en solución al 1% ( RE).
-Glicerina (PRS).
-Piedra pómez de 4-8 mm ( PR).
-Sodio hidróxido al 97% en lentejas (PA).
-Sodio hidróxido 0,1 N ( SV).
-Indicador de fenolftaleína.
La glicerina empleada tiene las siguientes características: d = 1,26; 98% (p/p).
Para preparar la solución acuosa de sodio hidróxido (44% p/p) se usa sodio hidróxido al 97% en lentejas (PA), y se disuelve en agua destilada (PA). Se recomienda su conservación en una botella protegida del dióxido de carbono, y utilizar siempre la porción limpia libre de precipitado de carbonatos.
El agua destilada (PA) que se utilice en este método debe hervirse durante 15 minutos, para eliminar el dióxido de carbono.
La solución acuosa de sodio hidróxido 0,1 N (SV) o potasio hidróxido 0,1 N, debe estar normalizada exactamente.
El alcohol etílico al 96% (v/v PA) debe ser neutro a la fenolftaleína; el agua usada debe ser destilada o de una pureza al menos equivalente.
Procedimiento analítico.
1.Preparación de la muestra: Fundir aproximadamente 50 g de la muestra de mantequilla en una estufa estándar a una temperatura inferior a 50 ºC, hasta conseguir la separación de las fases acuosa y grasa; seguidamente, hay que separar la capa de grasa por decantación, y clarificarla en la estufa a unos 40 ºC; después se filtra empleando un papel de filtro seco, evitando que pase la fase acuosa por el filtro.
2.Determinación del índice de ácidos grasos volátiles solubles: Pesar 5 g de grasa en el matraz A, con una aproximación de 0,01 g; añadir 20 g (16 ml) de glicerina (PRS), y 2 ml de solución de sodio hidróxido (44%). Para añadir la solución de sodio hidróxido debe usarse una bureta protegida de la entrada de dióxido de carbono, limpiando previamente la punta de la bureta y desechando las primeras gotas. Calentar el matraz a fuego directo, evitando el sobrecalentamiento, y agitando continuamente hasta que el líquido no forme espuma y se vuelva límpido. Dejar enfriar el matraz hasta 90 ºC, añadir 90 ml de agua destilada (PA) recién hervida a la misma temperatura aproximadamente, y mezclar hasta que el líquido obtenido sea límpido. Añadir de 0,6 a 0,7 g de piedra pómez y 50 ml de solución de ácido sulfúrico 1 N (SV). Conectar inmediatamente el matraz al aparato de destilación calentando ligeramente hasta que los ácidos grasos libres formen una capa superficial limpia. En el calentamiento debe regularse la llama de modo que se recojan en el matraz aforado 110 ml de destilado en unos 19-21 minutos, considerándose el comienzo de la destilación el momento en que se forma la primera gota en el refrigerante. Se debe regular el flujo de agua del refrigerante de modo que se mantenga la temperatura del agua que sale del mismo a 20 ± 1 ºC. Una vez recogidos los 110 ml de destilado, quitar el mechero inmediatamente y sustituir el matraz aforado por un pequeño vaso. A continuación, mezclar el contenido del matraz aforado agitando suavemente, y sumergir el matraz en un baño de agua a 20 ± 1 ºC durante 10-15 minutos, quedando la señal correspondiente a los 110 ml del matraz aforado por debajo del nivel del agua del baño; se debe agitar el matraz en varias ocasiones. Tapar el matraz y mezclar invirtiéndolo 4 o 5 veces sin agitar. Filtrar los 110 ml de destilado a través de un papel de filtro seco de velocidad media (diámetro 80-90 mm), que se ajusta en un embudo. El filtrado debe ser límpido, empleando un filtro con las dimensiones adecuadas para que un volumen de 15 ml lo llene por completo. Pipetar 100 ml de filtrado y pasarlos a un matraz erlenmeyer de 300 ml, añadir 0,5 ml de la solución indicadora de fenolftaleína al 1% (RE), y valorar con la solución acuosa de álcali “estándar” 0,1 N hasta obtener un color rosa persistente durante 30-60 segundos.
3.Ensayo en blanco: Se realiza sin grasa, y en lugar de saponificar a fuego directo hay que calentar en baño de agua hirviendo durante 15 minutos. Para la valoración es suficiente con emplear 0,5 ml de la solución de álcali normalizada. En caso contrario, se deben preparar nuevas soluciones del reactivo.
4.Determinación del índice de ácidos grasos volátiles insolubles (Polenske): Previamente, se debe lavar el filtro con tres volúmenes sucesivos de 15 ml de agua destilada (PA) a la temperatura de 20 ± 1 ºC, una vez que hayan pasado cada uno a través del refrigerante del vaso pequeño y del matraz aforado. Colocar el embudo y el filtro en el cuello de un matraz cónico, limpio y seco, de 200 ml de capacidad. Disolver los ácidos grasos insolubles repitiendo los lavados, usando volúmenes de 15 ml de alcohol etílico de 96% (v/v, PA). Valorar con la solución acuosa de álcali normalizada (0,1 N) el conjunto de los lavados utilizando alcohol etílico de 96% (v/v, PA), y 0,5 ml de solución indicadora de fenolftaleína al 1% (RE), hasta un color rosa persistente durante 30-60 segundos.
Expresión de los resultados.
Los distintos resultados se obtienen mediante las siguientes fórmulas:
1.Índice de ácidos grasos volátiles solubles (índice de Reichert):
Índice de Reichert = 11. t . (V1 – b)
teniendo en cuenta que el significado de los factores de la fórmula son:
V1 = Volumen en mililitros de la solución normalizada (0,1 N) de álcali, utilizados en la valoración de la muestra.
b = Volumen en mililitros de la solución normalizada (0,1 N) de álcali, utilizados en el ensayo en blanco.
t = Normalidad exacta de la solución normalizada (0,1 N) de álcali.
El resultado obtenido se redondea a la primera cifra decimal.
2.Índice de ácidos grasos volátiles insolubles (índice de Polenske):
Índice de Polenske = 10 . t . V2
teniendo en cuenta que el significado de los factores de la fórmula son:
V2 = Volumen en mililitros de la solución normalizada (0,1 N) de álcali utilizada en la valoración de la muestra.
t = Normalidad exacta de la solución normalizada (0,1 N) de álcali.
Se debe redondear el resultado a la primera cifra decimal.
3.Reproducción de los resultados: La diferencia entre los resultados de dos determinaciones duplicadas, obtenidos simultáneamente o inmediatamente uno detrás de otro por el mismo analista, no debe exceder de 0,5 para el índice de Reichert o de 0,3 para el índice de Polenske.
Referencias.

-Ácido sulfúrico 1 N (SV).
-Agua destilada (PA).
-Alcohol etílico al 96% (v/v, PA).
-Fenolftaleína en solución al 1% ( RE).
-Glicerina (PRS).
-Piedra pómez de 4-8 mm ( PR).
-Sodio hidróxido al 97% en lentejas (PA).
-Sodio hidróxido 0,1 N ( SV).
-Indicador de fenolftaleína.
La glicerina empleada tiene las siguientes características: d = 1,26; 98% (p/p).
Para preparar la solución acuosa de sodio hidróxido (44% p/p) se usa sodio hidróxido al 97% en lentejas (PA), y se disuelve en agua destilada (PA). Se recomienda su conservación en una botella protegida del dióxido de carbono, y utilizar siempre la porción limpia libre de precipitado de carbonatos.
El agua destilada (PA) que se utilice en este método debe hervirse durante 15 minutos, para eliminar el dióxido de carbono.
La solución acuosa de sodio hidróxido 0,1 N (SV) o potasio hidróxido 0,1 N, debe estar normalizada exactamente.
El alcohol etílico al 96% (v/v PA) debe ser neutro a la fenolftaleína; el agua usada debe ser destilada o de una pureza al menos equivalente.
Procedimiento analítico.
1.Preparación de la muestra: Fundir aproximadamente 50 g de la muestra de mantequilla en una estufa estándar a una temperatura inferior a 50 ºC, hasta conseguir la separación de las fases acuosa y grasa; seguidamente, hay que separar la capa de grasa por decantación, y clarificarla en la estufa a unos 40 ºC; después se filtra empleando un papel de filtro seco, evitando que pase la fase acuosa por el filtro.
2.Determinación del índice de ácidos grasos volátiles solubles: Pesar 5 g de grasa en el matraz A, con una aproximación de 0,01 g; añadir 20 g (16 ml) de glicerina (PRS), y 2 ml de solución de sodio hidróxido (44%). Para añadir la solución de sodio hidróxido debe usarse una bureta protegida de la entrada de dióxido de carbono, limpiando previamente la punta de la bureta y desechando las primeras gotas. Calentar el matraz a fuego directo, evitando el sobrecalentamiento, y agitando continuamente hasta que el líquido no forme espuma y se vuelva límpido. Dejar enfriar el matraz hasta 90 ºC, añadir 90 ml de agua destilada (PA) recién hervida a la misma temperatura aproximadamente, y mezclar hasta que el líquido obtenido sea límpido. Añadir de 0,6 a 0,7 g de piedra pómez y 50 ml de solución de ácido sulfúrico 1 N (SV). Conectar inmediatamente el matraz al aparato de destilación calentando ligeramente hasta que los ácidos grasos libres formen una capa superficial limpia. En el calentamiento debe regularse la llama de modo que se recojan en el matraz aforado 110 ml de destilado en unos 19-21 minutos, considerándose el comienzo de la destilación el momento en que se forma la primera gota en el refrigerante. Se debe regular el flujo de agua del refrigerante de modo que se mantenga la temperatura del agua que sale del mismo a 20 ± 1 ºC. Una vez recogidos los 110 ml de destilado, quitar el mechero inmediatamente y sustituir el matraz aforado por un pequeño vaso. A continuación, mezclar el contenido del matraz aforado agitando suavemente, y sumergir el matraz en un baño de agua a 20 ± 1 ºC durante 10-15 minutos, quedando la señal correspondiente a los 110 ml del matraz aforado por debajo del nivel del agua del baño; se debe agitar el matraz en varias ocasiones. Tapar el matraz y mezclar invirtiéndolo 4 o 5 veces sin agitar. Filtrar los 110 ml de destilado a través de un papel de filtro seco de velocidad media (diámetro 80-90 mm), que se ajusta en un embudo. El filtrado debe ser límpido, empleando un filtro con las dimensiones adecuadas para que un volumen de 15 ml lo llene por completo. Pipetar 100 ml de filtrado y pasarlos a un matraz erlenmeyer de 300 ml, añadir 0,5 ml de la solución indicadora de fenolftaleína al 1% (RE), y valorar con la solución acuosa de álcali “estándar” 0,1 N hasta obtener un color rosa persistente durante 30-60 segundos.
3.Ensayo en blanco: Se realiza sin grasa, y en lugar de saponificar a fuego directo hay que calentar en baño de agua hirviendo durante 15 minutos. Para la valoración es suficiente con emplear 0,5 ml de la solución de álcali normalizada. En caso contrario, se deben preparar nuevas soluciones del reactivo.
4.Determinación del índice de ácidos grasos volátiles insolubles (Polenske): Previamente, se debe lavar el filtro con tres volúmenes sucesivos de 15 ml de agua destilada (PA) a la temperatura de 20 ± 1 ºC, una vez que hayan pasado cada uno a través del refrigerante del vaso pequeño y del matraz aforado. Colocar el embudo y el filtro en el cuello de un matraz cónico, limpio y seco, de 200 ml de capacidad. Disolver los ácidos grasos insolubles repitiendo los lavados, usando volúmenes de 15 ml de alcohol etílico de 96% (v/v, PA). Valorar con la solución acuosa de álcali normalizada (0,1 N) el conjunto de los lavados utilizando alcohol etílico de 96% (v/v, PA), y 0,5 ml de solución indicadora de fenolftaleína al 1% (RE), hasta un color rosa persistente durante 30-60 segundos.
Expresión de los resultados.
Los distintos resultados se obtienen mediante las siguientes fórmulas:
1.Índice de ácidos grasos volátiles solubles (índice de Reichert):
Índice de Reichert = 11. t . (V1 – b)
teniendo en cuenta que el significado de los factores de la fórmula son:
V1 = Volumen en mililitros de la solución normalizada (0,1 N) de álcali, utilizados en la valoración de la muestra.
b = Volumen en mililitros de la solución normalizada (0,1 N) de álcali, utilizados en el ensayo en blanco.
t = Normalidad exacta de la solución normalizada (0,1 N) de álcali.
El resultado obtenido se redondea a la primera cifra decimal.
2.Índice de ácidos grasos volátiles insolubles (índice de Polenske):
Índice de Polenske = 10 . t . V2
teniendo en cuenta que el significado de los factores de la fórmula son:
V2 = Volumen en mililitros de la solución normalizada (0,1 N) de álcali utilizada en la valoración de la muestra.
t = Normalidad exacta de la solución normalizada (0,1 N) de álcali.
Se debe redondear el resultado a la primera cifra decimal.
3.Reproducción de los resultados: La diferencia entre los resultados de dos determinaciones duplicadas, obtenidos simultáneamente o inmediatamente uno detrás de otro por el mismo analista, no debe exceder de 0,5 para el índice de Reichert o de 0,3 para el índice de Polenske.
Referencias.
-Métodos de análisis. Boletín Oficial del Estado (21/7/1977 y 22/7/1977).
-Norma Internacional FIL-IDF 37: 1966.
-Métodos Oficiales de Análisis en Alimentaria: Leche y Productos Lácteos. Montplet & Esteban, 1987.
-Norma Internacional FIL-IDF 37: 1966.
-Métodos Oficiales de Análisis en Alimentaria: Leche y Productos Lácteos. Montplet & Esteban, 1987.
José Luis Ares Cea (coordinador de la Planta Piloto de Lácteos, Consejería de Agricultura y Pesca)