Continuando con los métodos oficiales de análisis de los productos lácteos, se expone seguidamente la metodología analítica de la determinación de la sacarosa en los helados.
Principios y fundamentos metodológicos.
Este método se basa en el principio de inversión de Clerget, que consiste en un tratamiento suave con ácido que hidroliza completamente la sacarosa. La lactosa y los otros azúcares prácticamente no se hidrolizan. La cantidad de sacarosa se deduce del cambio de poder rotatorio de la solución.
Se prepara un filtrado límpido de la muestra, sin mutarrotación debida a la lactosa, por tratamiento de la solución con amoníaco seguido de neutralización y clarificación por adiciones sucesivas de acetato de cinc y de ferrocianuro potásico. En una parte del filtrado la sacarosa se hidroliza en las condiciones especiales que corresponde a este tipo de operación. Partiendo de los poderes rotatorios del filtrado, antes y después de la inversión, se calcula la cantidad de sacarosa.
Material y aparatos utilizados.
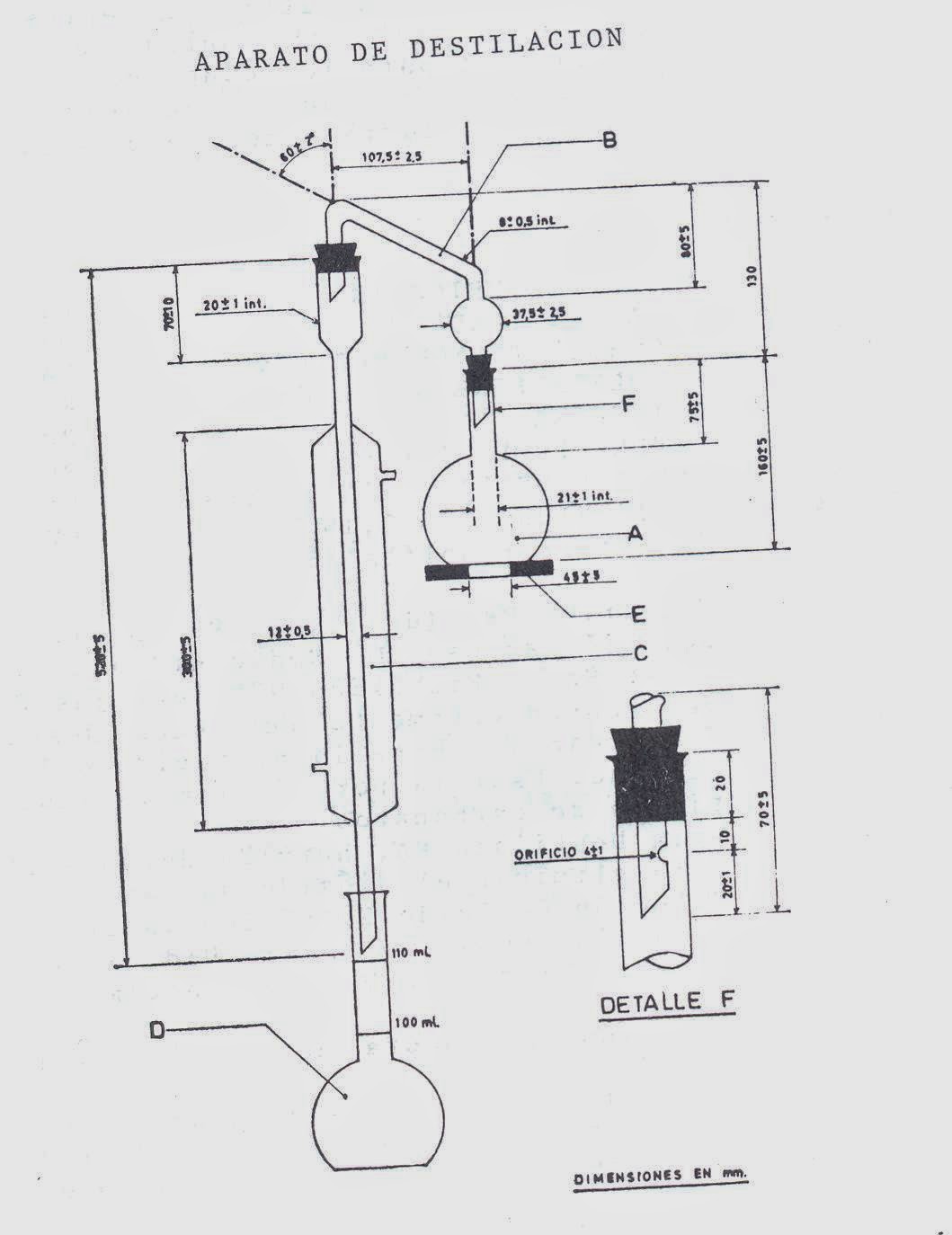
-Balanza analítica de sensibilidad 10 miligramos como mínimo.
-Vasos de precipitado, de 100 mililitros, de vidrio.
-Matraces graduados, de 200 y 50 mililitros.
-Pipetas, de 40 mililitros.
-Probetas graduadas, de 25 mililitros.
-Pipetas graduadas, de 10 militros.
-Embudo filtrante de diámetro entre 8 y 10 cm, y filtros (plegados) de 15 cm de diámetro.
-Tubo de polarímetro de 2 decímetros de longitud, exactamente calibrado.
-Polarímetro o sacarímetro.
-Polarímetro con luz de sodio o con luz verde de mercurio (lámpara de vapor de mercurio con prisma o pantalla Wratten nº 77A) permitiendo lecturas con una precisión por lo menos igual a 0,05 grados de ángulo.
-Sacarímetro con escala internacional de azúcar utilizando luz blanca, que pasa a través de un filtro de 15 milímetros de una solución al 6% de dicromato potásico, o bien luz de sodio, y permitiendo la lectura con una precisión por lo menos igual a 0,1 grados de la escala sacarimétrica internacional.
-Baño de agua a 60 ºC ± 1 ºC.
Reactivos necesarios.
-Acetato de cinc cristalizado: (C2H3O2)2Zn.2H2O.
-Ácido acético glacial al 96%.
-Agua destilada.
-Ferrocianuro potásico cristalizado: Fe(CN)6K4.3H2O.
-Ácido clorhídrico fumante al 37%.
-Amoníaco al 25%.
Preparación de la solución de acetato de cinc 2,0 N: Disolver 21,9 g de acetato de cinc cristalizado en 3 ml de agua destilada, completando el volumen hasta 100 ml (enrase).
Preparación de la solución de ferrocianuro potásico 1,0 N: Disolver 10,6 g de ferrocianuro potásico cristalizado en agua destilada, y completar hasta 100 ml.
Preparación de la solución de ácido clorhídrico 6,35 ± 0,20 N (20-22%): Diluir 52,5 ml de ácido clorhídrico fumante (37%) en agua destilada, y completar hasta 100 ml.
Preparación de la solución diluida de amoníaco 2,0 ± 0,2 N (3,5%): Diluir 15,2 ml de solución de amoníaco (25%) en agua destilada, y completar hasta 100 ml.
Preparación de la solución diluida ácido acético 2,0 ± 0,20 N (12%): Diluir 11,8 ml de ácido acético glacial (96%) en agua destilada, y completar hasta 100 ml.
Procedimiento analítico.
1. Acondicionamiento de las muestras:
a) Para muestras de productos recientemente preparados en los que no se pueda prever separación alguna apreciable de los componentes:
Abrir el recipiente, introducir en él el producto adherido a la tapa y mediante un movimiento de arriba-abajo, con ayuda de una cuchara, conseguir que se mezclen íntimamente las capas superiores, así como el contenido del fondo del recipiente. Trasvasar el contenido del bote a un frasco provisto de tapón bien adaptado.
b) Para muestras de productos más antiguos y muestras en las que se pueda prever una separación de componentes:
Calentar en baño de agua, aproximadamente a 40 ºC, hasta que la muestra casi haya alcanzado esta temperatura, abrir el recipiente y operar de la misma manera que en el punto a). En el caso de emplear un bote, hay que trasvasar su contenido a un frasco, raspar el producto que se haya adherido a las paredes y continuar la mezcla hasta que toda la masa sea homogénea. Cerrar el frasco con una tapadera que se adapte perfectamente. Dejar enfriar.
2. Comprobación del método:
Proceder como en el apartado 4.3, utilizando una mezcla de 100 g de leche o de 110 g de leche desnatada, y de 18 g de sacarosa pura, que corresponde a 40 g de una leche concentrada conteniendo el 45% de sacarosa. Calcular la cantidad de sacarosa como en el apartado 5.1, utilizando la fórmula (I), para W, F y P, la cantidad de leche pesada y la riqueza en materia grasa y proteínas de la leche, respectivamente; en la fórmula (II), para W, se utiliza la cifra de 40,00. Finalmente, se comprueba que la media de los valores encontrados no difiera de dicho valor (45%) en más de 0,1%.
3. Determinación:
3.1. En un vaso de 100 ml pesar aproximadamente 40 g de mezcla bien mezclada con una aproximación de 10 mg, y añadir 50 ml de agua destilada caliente (80-90 ºC) y mezclar cuidadosamente. Trasvasar cuantitativamente la mezcla a un matraz aforado de 200 ml, enjuagar el vaso con cantidades sucesivas de agua destilada a 60 ºC hasta alcanzar un volumen total de 120-150 ml. Mezclar y enfriar a temperatura ambiente. Añadir 5 ml de la solución de amoníaco diluida. Mezclar nuevamente y dejar reposar durante 15 minutos. Neutralizar el amoníaco añadiendo una cantidad equivalente de la solución diluida en ácido acético. Determinar previamente el volumen exacto (en ml) mediante la valoración de la solución de amoníaco diluida empleando el azul de bromotimol como indicador. Seguidamente se mezcla y se añade 12,5 ml de la solución de acetato, mezclando suavemente por rotación del matraz inclinado. Añadir 12,5 ml de la solución de ferrocianuro potásico. El contenido del matraz se pone a la temperatura de 20 ºC, y se añade agua destilada (a 20 ºC) hasta alcanzar el enrase de 200 mililitros. Tapar el matraz con un tapón seco y mezclar íntimamente sacudiendo con energía. Dejar reposar durante algunos minutos, filtrar a continuación por un papel de filtro seco, desechando los primeros 25 ml del filtrado.
Recomendación: Todas las adiciones de agua o de reactivos deberán hacerse de tal manera que se evite la formación de burbujas de aire; por este motivo, todas las mezclas deberán realizarse por rotación y no por agitación violenta. En el caso de observar la presencia de burbujas de aire antes de alcanzar los 200 ml deberán eliminarse aplicando al matraz una bomba de vacío e imprimiendo un movimiento de rotación.
3.2. Polarización directa: Determinar la rotación óptica del filtrado a 20 ±2 ºC.
3.3. Inversión: Introducir con la pipeta en un matraz graduado de 50 ml, 40 ml del filtrado obtenido de la manera indicada anteriormente. Añadir 6,0 ml de ácido clorhídrico 6,35 N. Poner el matraz en baño de agua a 60 ºC durante 15 minutos, sumergiéndolo hasta el nacimiento del cuello del mismo. Mezclar por rotación durante los 5 primeros minutos, alcanzando el contenido la temperatura del baño de agua. Enfriar a 20 ºC, y completar hasta 50 ml con agua destilada a 20 ºC. Mezclar y dejar reposar a esta temperatura durante 60 minutos.
3.4. Polarización después de la inversión: Determinar el poder rotatorio de la solución invertida a 20 ± 2 ºC. En el caso de que la temperatura del líquido en el tubo de polarización difiera en más de 0,2 ºC del valor prefijado durante la medida (20 ºC), deberá aplicarse la corrección de la temperatura indicada en el apartado 5.2.
Expresión de los resultados.
Los distintos resultados se obtienen mediante las siguientes fórmulas:
1. Riqueza en sacarosa:
(I) υ= (1,08 F + 1,55 P) W/100
(II) S = D - l 5/4/ Q x V – v/ V x v/L.W x 100
siendo:
S = Cantidad de sacarosa.
W = Peso de la muestra expresado en gramos.
P = Porcentaje de proteínas (N x 6,38) de la muestra.
F = Porcentaje en materia grasa de la muestra.
V = Volumen en mililitros de la muestra diluida antes de filtrar.
D = Lectura polarimétrica directa (polarización antes de la inversión).
l = Lectura polirimétrica después de la inversión.
L = Longitud en decímetros del tubo del polarímetro.
Q = Factor de inversión cuyos valores se indican más adelante.
Pesando exactamente 40,00 g de leche condensada y utilizando un polarímetro con luz de sodio provisto de escala en grados de ángulo y un tubo de 2 decímetros de longitud, el contenido en sacarosa de las leches condensadas normales (C=9) a 20,0 ± 1 ºC, se puede calcular con la siguiente fórmula:
S = (D – 5/4 I) x (2,833 – 0,00612 F – 0,00878 P)
Cuando la medida de la polarización después de la inversión se efectúa a una temperatura diferente de 20 ºC, las cifras obtenidas se deben multiplicar por la siguiente expresión: [(1 + 0,0037 (T – 20)].
2. Valores del factor de inversión Q:
Las fórmulas siguientes dan valores precisos de Q para diversas clases de luz con correcciones, si es necesario, para la concentración y la temperatura.
-Luz de sodio y polarímetro con escala en grados de ángulo:
Q = 0,8825 + 0,0006 (C-9) – 0,003 (T-20)
-Luz verde de mercurio y el polarímetro con escala de ángulo:
Q = 1,0392 + 0,0007 (C – 9) – 0,0039 (T-20)
-Luz blanca con filtro de dicromato y sacarímetro con escala sacarimétrica:
Q = 2,549 + 0,0017 (C – 9) – 0,0095 (T-20)
La nomenclatura de las fórmulas precedentes es:
C = Porcentaje de azúcares totales en la solución invertida, según la lectura polarimétrica.
T = Temperatura de la solución invertida en la lectura del polarímetro.
El porcentaje de azúcaes totales (C) en la solución invertida se puede calcular a partir de la lectura directa y de la variación después de la inversión según el método habitual, utilizando los valores usuales de rotación específica de sacarosa, de lactosa y de sacarosa invertida. La corrección 0,0006 (C-9), etc., no es exacta más que cuando C es aproximadamente 9; para la leche concentrada normal esta corrección se puede despreciar, por ser C próximo a 9.
Las variaciones en la temperatura del valor de 20 ºC influyen escasamente en la lectura de la polarización directa. Por el contrario, diferencias de más de 0,2 ºC en la lectura de polarización después de la inversión necesitan una corrección. La corrección -0,003 (T-20), etc., no es exacta más que para temperaturas comprendidas entre 18 ºC y 22 ºC.
La diferencia entre los resultados de dos determinaciones efectuadas simultáneamente o de una inmediatamente después de otra, por el mismo analista, no debe ser mayor de 0,3 gramos de sacarosa por 100 gramos de leche concentrada.
Referencias.
Principios y fundamentos metodológicos.
Este método se basa en el principio de inversión de Clerget, que consiste en un tratamiento suave con ácido que hidroliza completamente la sacarosa. La lactosa y los otros azúcares prácticamente no se hidrolizan. La cantidad de sacarosa se deduce del cambio de poder rotatorio de la solución.
Se prepara un filtrado límpido de la muestra, sin mutarrotación debida a la lactosa, por tratamiento de la solución con amoníaco seguido de neutralización y clarificación por adiciones sucesivas de acetato de cinc y de ferrocianuro potásico. En una parte del filtrado la sacarosa se hidroliza en las condiciones especiales que corresponde a este tipo de operación. Partiendo de los poderes rotatorios del filtrado, antes y después de la inversión, se calcula la cantidad de sacarosa.
Material y aparatos utilizados.
-Balanza analítica de sensibilidad 10 miligramos como mínimo.
-Vasos de precipitado, de 100 mililitros, de vidrio.
-Matraces graduados, de 200 y 50 mililitros.
-Pipetas, de 40 mililitros.
-Probetas graduadas, de 25 mililitros.
-Pipetas graduadas, de 10 militros.
-Embudo filtrante de diámetro entre 8 y 10 cm, y filtros (plegados) de 15 cm de diámetro.
-Tubo de polarímetro de 2 decímetros de longitud, exactamente calibrado.
-Polarímetro o sacarímetro.
-Polarímetro con luz de sodio o con luz verde de mercurio (lámpara de vapor de mercurio con prisma o pantalla Wratten nº 77A) permitiendo lecturas con una precisión por lo menos igual a 0,05 grados de ángulo.
-Sacarímetro con escala internacional de azúcar utilizando luz blanca, que pasa a través de un filtro de 15 milímetros de una solución al 6% de dicromato potásico, o bien luz de sodio, y permitiendo la lectura con una precisión por lo menos igual a 0,1 grados de la escala sacarimétrica internacional.
-Baño de agua a 60 ºC ± 1 ºC.
Reactivos necesarios.
-Acetato de cinc cristalizado: (C2H3O2)2Zn.2H2O.
-Ácido acético glacial al 96%.
-Agua destilada.
-Ferrocianuro potásico cristalizado: Fe(CN)6K4.3H2O.
-Ácido clorhídrico fumante al 37%.
-Amoníaco al 25%.
Preparación de la solución de acetato de cinc 2,0 N: Disolver 21,9 g de acetato de cinc cristalizado en 3 ml de agua destilada, completando el volumen hasta 100 ml (enrase).
Preparación de la solución de ferrocianuro potásico 1,0 N: Disolver 10,6 g de ferrocianuro potásico cristalizado en agua destilada, y completar hasta 100 ml.
Preparación de la solución de ácido clorhídrico 6,35 ± 0,20 N (20-22%): Diluir 52,5 ml de ácido clorhídrico fumante (37%) en agua destilada, y completar hasta 100 ml.
Preparación de la solución diluida de amoníaco 2,0 ± 0,2 N (3,5%): Diluir 15,2 ml de solución de amoníaco (25%) en agua destilada, y completar hasta 100 ml.
Preparación de la solución diluida ácido acético 2,0 ± 0,20 N (12%): Diluir 11,8 ml de ácido acético glacial (96%) en agua destilada, y completar hasta 100 ml.
Procedimiento analítico.
1. Acondicionamiento de las muestras:
a) Para muestras de productos recientemente preparados en los que no se pueda prever separación alguna apreciable de los componentes:
Abrir el recipiente, introducir en él el producto adherido a la tapa y mediante un movimiento de arriba-abajo, con ayuda de una cuchara, conseguir que se mezclen íntimamente las capas superiores, así como el contenido del fondo del recipiente. Trasvasar el contenido del bote a un frasco provisto de tapón bien adaptado.
b) Para muestras de productos más antiguos y muestras en las que se pueda prever una separación de componentes:
Calentar en baño de agua, aproximadamente a 40 ºC, hasta que la muestra casi haya alcanzado esta temperatura, abrir el recipiente y operar de la misma manera que en el punto a). En el caso de emplear un bote, hay que trasvasar su contenido a un frasco, raspar el producto que se haya adherido a las paredes y continuar la mezcla hasta que toda la masa sea homogénea. Cerrar el frasco con una tapadera que se adapte perfectamente. Dejar enfriar.
2. Comprobación del método:
Proceder como en el apartado 4.3, utilizando una mezcla de 100 g de leche o de 110 g de leche desnatada, y de 18 g de sacarosa pura, que corresponde a 40 g de una leche concentrada conteniendo el 45% de sacarosa. Calcular la cantidad de sacarosa como en el apartado 5.1, utilizando la fórmula (I), para W, F y P, la cantidad de leche pesada y la riqueza en materia grasa y proteínas de la leche, respectivamente; en la fórmula (II), para W, se utiliza la cifra de 40,00. Finalmente, se comprueba que la media de los valores encontrados no difiera de dicho valor (45%) en más de 0,1%.
3. Determinación:
3.1. En un vaso de 100 ml pesar aproximadamente 40 g de mezcla bien mezclada con una aproximación de 10 mg, y añadir 50 ml de agua destilada caliente (80-90 ºC) y mezclar cuidadosamente. Trasvasar cuantitativamente la mezcla a un matraz aforado de 200 ml, enjuagar el vaso con cantidades sucesivas de agua destilada a 60 ºC hasta alcanzar un volumen total de 120-150 ml. Mezclar y enfriar a temperatura ambiente. Añadir 5 ml de la solución de amoníaco diluida. Mezclar nuevamente y dejar reposar durante 15 minutos. Neutralizar el amoníaco añadiendo una cantidad equivalente de la solución diluida en ácido acético. Determinar previamente el volumen exacto (en ml) mediante la valoración de la solución de amoníaco diluida empleando el azul de bromotimol como indicador. Seguidamente se mezcla y se añade 12,5 ml de la solución de acetato, mezclando suavemente por rotación del matraz inclinado. Añadir 12,5 ml de la solución de ferrocianuro potásico. El contenido del matraz se pone a la temperatura de 20 ºC, y se añade agua destilada (a 20 ºC) hasta alcanzar el enrase de 200 mililitros. Tapar el matraz con un tapón seco y mezclar íntimamente sacudiendo con energía. Dejar reposar durante algunos minutos, filtrar a continuación por un papel de filtro seco, desechando los primeros 25 ml del filtrado.
Recomendación: Todas las adiciones de agua o de reactivos deberán hacerse de tal manera que se evite la formación de burbujas de aire; por este motivo, todas las mezclas deberán realizarse por rotación y no por agitación violenta. En el caso de observar la presencia de burbujas de aire antes de alcanzar los 200 ml deberán eliminarse aplicando al matraz una bomba de vacío e imprimiendo un movimiento de rotación.
3.2. Polarización directa: Determinar la rotación óptica del filtrado a 20 ±2 ºC.
3.3. Inversión: Introducir con la pipeta en un matraz graduado de 50 ml, 40 ml del filtrado obtenido de la manera indicada anteriormente. Añadir 6,0 ml de ácido clorhídrico 6,35 N. Poner el matraz en baño de agua a 60 ºC durante 15 minutos, sumergiéndolo hasta el nacimiento del cuello del mismo. Mezclar por rotación durante los 5 primeros minutos, alcanzando el contenido la temperatura del baño de agua. Enfriar a 20 ºC, y completar hasta 50 ml con agua destilada a 20 ºC. Mezclar y dejar reposar a esta temperatura durante 60 minutos.
3.4. Polarización después de la inversión: Determinar el poder rotatorio de la solución invertida a 20 ± 2 ºC. En el caso de que la temperatura del líquido en el tubo de polarización difiera en más de 0,2 ºC del valor prefijado durante la medida (20 ºC), deberá aplicarse la corrección de la temperatura indicada en el apartado 5.2.
Expresión de los resultados.
Los distintos resultados se obtienen mediante las siguientes fórmulas:
1. Riqueza en sacarosa:
(I) υ= (1,08 F + 1,55 P) W/100
(II) S = D - l 5/4/ Q x V – v/ V x v/L.W x 100
siendo:
S = Cantidad de sacarosa.
W = Peso de la muestra expresado en gramos.
P = Porcentaje de proteínas (N x 6,38) de la muestra.
F = Porcentaje en materia grasa de la muestra.
V = Volumen en mililitros de la muestra diluida antes de filtrar.
D = Lectura polarimétrica directa (polarización antes de la inversión).
l = Lectura polirimétrica después de la inversión.
L = Longitud en decímetros del tubo del polarímetro.
Q = Factor de inversión cuyos valores se indican más adelante.
Pesando exactamente 40,00 g de leche condensada y utilizando un polarímetro con luz de sodio provisto de escala en grados de ángulo y un tubo de 2 decímetros de longitud, el contenido en sacarosa de las leches condensadas normales (C=9) a 20,0 ± 1 ºC, se puede calcular con la siguiente fórmula:
S = (D – 5/4 I) x (2,833 – 0,00612 F – 0,00878 P)
Cuando la medida de la polarización después de la inversión se efectúa a una temperatura diferente de 20 ºC, las cifras obtenidas se deben multiplicar por la siguiente expresión: [(1 + 0,0037 (T – 20)].
2. Valores del factor de inversión Q:
Las fórmulas siguientes dan valores precisos de Q para diversas clases de luz con correcciones, si es necesario, para la concentración y la temperatura.
-Luz de sodio y polarímetro con escala en grados de ángulo:
Q = 0,8825 + 0,0006 (C-9) – 0,003 (T-20)
-Luz verde de mercurio y el polarímetro con escala de ángulo:
Q = 1,0392 + 0,0007 (C – 9) – 0,0039 (T-20)
-Luz blanca con filtro de dicromato y sacarímetro con escala sacarimétrica:
Q = 2,549 + 0,0017 (C – 9) – 0,0095 (T-20)
La nomenclatura de las fórmulas precedentes es:
C = Porcentaje de azúcares totales en la solución invertida, según la lectura polarimétrica.
T = Temperatura de la solución invertida en la lectura del polarímetro.
El porcentaje de azúcaes totales (C) en la solución invertida se puede calcular a partir de la lectura directa y de la variación después de la inversión según el método habitual, utilizando los valores usuales de rotación específica de sacarosa, de lactosa y de sacarosa invertida. La corrección 0,0006 (C-9), etc., no es exacta más que cuando C es aproximadamente 9; para la leche concentrada normal esta corrección se puede despreciar, por ser C próximo a 9.
Las variaciones en la temperatura del valor de 20 ºC influyen escasamente en la lectura de la polarización directa. Por el contrario, diferencias de más de 0,2 ºC en la lectura de polarización después de la inversión necesitan una corrección. La corrección -0,003 (T-20), etc., no es exacta más que para temperaturas comprendidas entre 18 ºC y 22 ºC.
La diferencia entre los resultados de dos determinaciones efectuadas simultáneamente o de una inmediatamente después de otra, por el mismo analista, no debe ser mayor de 0,3 gramos de sacarosa por 100 gramos de leche concentrada.
Referencias.
-Métodos de Análisis Lactológicos. Industrias Lácteas Españolas, 1982.

José Luis Ares Cea (coordinador de la Planta Piloto de Lácteos, Consejería de Agricultura y Pesca)
José Luis Ares Cea (coordinador de la Planta Piloto de Lácteos, Consejería de Agricultura y Pesca)